


 Sign Out
Sign Out
Pharmacodynamics: Risperidone is a monoaminergic antagonist with high affinity (Ki of 0.12 to 7.3 nM) for the serotonin Type 2 (5HT2), dopamine Type 2 (D2), α1 and α2 adrenergic, and H1 histaminergic receptors. Risperidone showed low to moderate affinity (Ki of 47 to 253 nM) for the serotonin 5HT1C, 5HT1D, and 5HT1A receptors, weak affinity (Ki of 620 to 800 nM) for the dopamine D1 and haloperidol-sensitive sigma site, and no affinity (when tested at concentrations >10-5 M) for cholinergic muscarinic or β1 and β2 adrenergic receptors.
Clinical Studies: Risperdal: Schizophrenia: Adults: Short-Term Efficacy: The efficacy of RISPERDAL in the treatment of schizophrenia was established in four short-term (4- to 8-week) controlled trials of psychotic inpatients who met DSM‑III-R criteria for schizophrenia.
Several instruments were used for assessing psychiatric signs and symptoms in these studies, among them the Brief Psychiatric Rating Scale (BPRS), a multi-item inventory of general psychopathology traditionally used to evaluate the effects of drug treatment in schizophrenia. The BPRS psychosis cluster (conceptual disorganization, hallucinatory behavior, suspiciousness, and unusual thought content) is considered a particularly useful subset for assessing actively psychotic schizophrenic patients. A second traditional assessment, the Clinical Global Impression (CGI), reflects the impression of a skilled observer, fully familiar with the manifestations of schizophrenia, about the overall clinical state of the patient. In addition, the Positive and Negative Syndrome Scale (PANSS) and the Scale for Assessing Negative Symptoms (SANS) were employed.
The results of the trials follow: (1) In a 6-week, placebo-controlled trial (n=160) involving titration of RISPERDAL in doses up to 10 mg/day (twice-daily schedule), RISPERDAL was generally superior to placebo on the BPRS total score, on the BPRS psychosis cluster, and marginally superior to placebo on the SANS.
(2) In an 8-week, placebo-controlled trial (n=513) involving 4 fixed doses of RISPERDAL (2 mg/day, 6 mg/day, 10 mg/day, and 16 mg/day, on a twice-daily schedule), all 4 RISPERDAL groups were generally superior to placebo on the BPRS total score, BPRS psychosis cluster, and CGI severity score; the 3 highest RISPERDAL dose groups were generally superior to placebo on the PANSS negative subscale. The most consistently positive responses on all measures were seen for the 6 mg dose group, and there was no suggestion of increased benefit from larger doses.
(3) In an 8-week, dose comparison trial (n=1356) involving 5 fixed doses of RISPERDAL (1 mg/day, 4 mg/day, 8 mg/day, 12 mg/day, and 16 mg/day, on a twice-daily schedule), the four highest RISPERDAL dose groups were generally superior to the 1 mg RISPERDAL dose group on BPRS total score, BPRS psychosis cluster, and CGI severity score. None of the dose groups were superior to the 1 mg group on the PANSS negative subscale. The most consistently positive responses were seen for the 4 mg dose group.
(4) In a 4-week, placebo-controlled dose comparison trial (n=246) involving 2 fixed doses of RISPERDAL (4 and 8 mg/day on a once-daily schedule), both RISPERDAL dose groups were generally superior to placebo on several PANSS measures, including a response measure (>20% reduction in PANSS total score), PANSS total score, and the BPRS psychosis cluster (derived from PANSS). The results were generally stronger for the 8 mg than for the 4 mg dose group.
Long-Term Efficacy: In a longer-term trial, 365 adult outpatients predominantly meeting DSM-IV criteria for schizophrenia and who had been clinically stable for at least 4 weeks on an antipsychotic medication were randomized to RISPERDAL (2-8 mg/day) or to an active comparator, for 1 to 2 years of observation for relapse. Patients receiving RISPERDAL experienced a significantly longer time to relapse over this time period compared to those receiving the active comparator.
Pediatrics: The efficacy of RISPERDAL in the treatment of schizophrenia in adolescents aged 13-17 years was demonstrated in two short-term (6 and 8 weeks), double-blind controlled trials. All patients met DSM-IV diagnostic criteria for schizophrenia and were experiencing an acute episode at time of enrollment. In the first trial (study #1), patients were randomized into one of three treatment groups: RISPERDAL 1-3 mg/day (n = 55, mean modal dose = 2.6 mg), RISPERDAL 4-6 mg/day (n = 51, mean modal dose = 5.3 mg), or placebo (n = 54). In the second trial (study #2), patients were randomized to either RISPERDAL 0.15‑0.6 mg/day (n = 132, mean modal dose = 0.5 mg) or RISPERDAL 1.5-6 mg/day (n = 125, mean modal dose = 4 mg). In all cases, study medication was initiated at 0.5 mg/day (with the exception of the 0.15-0.6 mg/day group in study #2, where the initial dose was 0.05 mg/day) and titrated to the target dosage range by approximately Day 7. Subsequently, dosage was increased to the maximum tolerated dose within the target dose range by Day 14. The primary efficacy variable in all studies was the mean change from baseline in total PANSS score.
Results of the studies demonstrated efficacy of RISPERDAL in all dose groups from 1‑6 mg/day compared to placebo, as measured by significant reduction of total PANSS score. The efficacy on the primary parameter in the 1-3 mg/day group was comparable to the 4‑6 mg/day group in study #1, and similar to the efficacy demonstrated in the 1.5-6 mg/day group in study #2. In study #2, the efficacy in the 1.5-6 mg/day group was statistically significantly greater than that in the 0.15-0.6 mg/day group. Doses higher than 3 mg/day did not reveal any trend towards greater efficacy.
Bipolar Mania - Monotherapy: Adults: The efficacy of RISPERDAL in the treatment of acute manic or mixed episodes was established in two short-term (3-week) placebo-controlled trials in patients who met the DSM-IV criteria for Bipolar I Disorder with manic or mixed episodes. These trials included patients with or without psychotic features.
The primary rating instrument used for assessing manic symptoms in these trials was the Young Mania Rating Scale (YMRS), an 11-item clinician-rated scale traditionally used to assess the degree of manic symptomatology (irritability, disruptive/aggressive behavior, sleep, elevated mood, speech, increased activity, sexual interest, language/thought disorder, thought content, appearance, and insight) in a range from 0 (no manic features) to 60 (maximum score). The primary outcome in these trials was change from baseline in the YMRS total score. The results of the trials follow: (1) In one 3-week placebo-controlled trial (n=246), limited to patients with manic episodes, which involved a dose range of RISPERDAL 1-6 mg/day, once daily, starting at 3 mg/day (mean modal dose was 4.1 mg/day), RISPERDAL was superior to placebo in the reduction of YMRS total score.
(2) In another 3-week placebo-controlled trial (n=286), which involved a dose range of 1‑6 mg/day, once daily, starting at 3 mg/day (mean modal dose was 5.6 mg/day), RISPERDAL was superior to placebo in the reduction of YMRS total score.
Pediatrics: The efficacy of RISPERDAL in the treatment of mania in children or adolescents with Bipolar I disorder was demonstrated in a 3-week, randomized, double-blind, placebo-controlled, multicenter trial including patients ranging in ages from 10 to 17 years who were experiencing a manic or mixed episode of bipolar I disorder. Patients were randomized into one of three treatment groups: RISPERDAL 0.5-2.5 mg/day (n = 50, mean modal dose = 1.9 mg), RISPERDAL 3-6 mg/day (n = 61, mean modal dose = 4.7 mg), or placebo (n = 58). In all cases, study medication was initiated at 0.5 mg/day and titrated to the target dosage range by Day 7, with further increases in dosage to the maximum tolerated dose within the targeted dose range by Day 10. The primary rating instrument used for assessing efficacy in this study was the mean change from baseline in the total YMRS score.
Results of this study demonstrated efficacy of RISPERDAL in both dose groups compared with placebo, as measured by significant reduction of total YMRS score. The efficacy on the primary parameter in the 3-6 mg/day dose group was comparable to the 0.5-2.5 mg/day dose group. Doses higher than 2.5 mg/day did not reveal any trend towards greater efficacy.
Bipolar Mania - Adjunctive Therapy with Lithium or Valproate: The efficacy of RISPERDAL with concomitant lithium or valproate in the treatment of acute manic or mixed episodes was established in one controlled trial in adult patients who met the DSM-IV criteria for Bipolar I Disorder. This trial included patients with or without psychotic features and with or without a rapid-cycling course.
(1) In this 3-week placebo-controlled combination trial, 148 in- or outpatients on lithium or valproate therapy with inadequately controlled manic or mixed symptoms were randomized to receive RISPERDAL, placebo, or an active comparator, in combination with their original therapy. RISPERDAL, in a dose range of 1-6 mg/day, once daily, starting at 2 mg/day (mean modal dose of 3.8 mg/day), combined with lithium or valproate (in a therapeutic range of 0.6 mEq/L to 1.4 mEq/L or 50 mcg/mL to 120 mcg/mL, respectively) was superior to lithium or valproate alone in the reduction of YMRS total score.
(2) In a second 3-week placebo-controlled combination trial, 142 in- or outpatients on lithium, valproate, or carbamazepine therapy with inadequately controlled manic or mixed symptoms were randomized to receive RISPERDAL or placebo, in combination with their original therapy. RISPERDAL, in a dose range of 1‑6 mg/day, once daily, starting at 2 mg/day (mean modal dose of 3.7 mg/day), combined with lithium, valproate, or carbamazepine (in therapeutic ranges of 0.6 mEq/L to 1.4 mEq/L for lithium, 50 mcg/mL to 125 mcg/mL for valproate, or 4-12 mcg/mL for carbamazepine, respectively) was not superior to lithium, valproate, or carbamazepine alone in the reduction of YMRS total score. A possible explanation for the failure of this trial was induction of risperidone and 9‑hydroxyrisperidone clearance by carbamazepine, leading to subtherapeutic levels of risperidone and 9‑hydroxyrisperidone.
Irritability Associated with Autistic Disorder: Short-Term Efficacy: The efficacy of RISPERDAL in the treatment of irritability associated with autistic disorder was established in two 8-week, placebo-controlled trials in children and adolescents (aged 5 to 16 years) who met the DSM-IV criteria for autistic disorder. Over 90% of these subjects were under 12 years of age and most weighed over 20 kg (16-104.3 kg).
Efficacy was evaluated using two assessment scales: the Aberrant Behavior Checklist (ABC) and the Clinical Global Impression - Change (CGI-C) scale. The primary outcome measure in both trials was the change from baseline to endpoint in the Irritability subscale of the ABC (ABC-I). The ABC-I subscale measured the emotional and behavioral symptoms of autism, including aggression towards others, deliberate self-injuriousness, temper tantrums, and quickly changing moods. The CGI-C rating at endpoint was a co-primary outcome measure in one of the studies.
The results of these trials are as follows: (1) In one of the 8-week, placebo-controlled trials, children and adolescents with autistic disorder (n=101), aged 5 to 16 years, received twice daily doses of placebo or RISPERDAL 0.5-3.5 mg/day on a weight‑adjusted basis. RISPERDAL, starting at 0.25 mg/day or 0.5 mg/day depending on baseline weight (< 20 kg and ≥ 20 kg, respectively) and titrated to clinical response (mean modal dose of 1.9 mg/day, equivalent to 0.06 mg/kg/day), significantly improved scores on the ABC-I subscale and on the CGI-C scale compared with placebo.
(2) In the other 8-week, placebo-controlled trial in children with autistic disorder (n=55), aged 5 to 12 years, RISPERDAL 0.02 to 0.06 mg/kg/day given once or twice daily, starting at 0.01 mg/kg/day and titrated to clinical response (mean modal dose of 0.05 mg/kg/day, equivalent to 1.4 mg/day), significantly improved scores on the ABC-I subscale compared with placebo.
A third trial was a 6-week, multicenter, randomized, double-blind, placebo-controlled, fixed-dose study to evaluate the efficacy and safety of a lower than recommended dose of risperidone in subjects (N=96) 5 to 17 years of age with autistic disorder (defined by DSM-IV criteria) and associated irritability and related behavioral symptoms. Approximately 77% of patients were younger than 12 years of age (mean age = 9), and 88% were male. Most patients (73%) weighed less than 45 kg (mean weight = 40 kg). Approximately 90% of patients were antipsychotic-naïve before entering the study.
There were two weight-based, fixed doses of risperidone (high-dose and low-dose). The high dose was 1.25 mg per day for patients weighing 20 to < 45 kg, and it was 1.75 mg per day for patients weighing > 45 kg. The low dose was 0.125 mg per day for patients weighing 20 to < 45 kg, and it was 0.175 mg per day for patients weighing > 45 kg. The dose was administered once daily in the morning, or in the evening if sedation occurred.
The primary efficacy endpoint was the mean change in the Aberrant Behavior Checklist - Irritability subscale (ABC-I) score from baseline to the end of Week 6. The study demonstrated the efficacy of high-dose risperidone, as measured by the mean change in ABC-I score. It did not demonstrate efficacy for low-dose risperidone. The mean baseline ABC-I scores were 29 in the placebo group (n = 35), 27 in the risperidone low-dose group (n = 30), and 28 in the risperidone high-dose group (n = 31). The mean changes in ABC-I scores were -3.5, -7.4, and -12.4 in the placebo, low-dose, and high-dose group respectively. The results in the high-dose group were statistically significant (p<0.001) but not in the low-dose group (p=0.164).
Long-Term Efficacy: Following completion of the first 8-week double-blind study, 63 patients entered an open-label study extension where they were treated with RISPERDAL for 4 or 6 months (depending on whether they received RISPERDAL or placebo in the double-blind study). During this open-label treatment period, patients were maintained on a mean modal dose of RISPERDAL of 1.8‑2.1 mg/day (equivalent to 0.05 - 0.07 mg/kg/day).
Patients who maintained their positive response to RISPERDAL (response was defined as ≥ 25% improvement on the ABC-I subscale and a CGI-C rating of 'much improved' or 'very much improved') during the 4-6 month open-label treatment phase for about 140 days, on average, were randomized to receive RISPERDAL or placebo during an 8‑week, double-blind withdrawal study (n=39 of the 63 patients). A pre-planned interim analysis of data from patients who completed the withdrawal study (n=32), undertaken by an independent Data Safety Monitoring Board, demonstrated a significantly lower relapse rate in the RISPERDAL group compared with the placebo group. Based on the interim analysis results, the study was terminated due to demonstration of a statistically significant effect on relapse prevention. Relapse was defined as ≥ 25% worsening on the most recent assessment of the ABC-I subscale (in relation to baseline of the randomized withdrawal phase).
Risperdal Consta: Schizophrenia: The effectiveness of RISPERDAL CONSTA in the treatment of schizophrenia was established, in part, on the basis of extrapolation from the established effectiveness of the oral formulation of risperidone. In addition, the effectiveness of RISPERDAL CONSTA in the treatment of schizophrenia was established in a 12-week, placebo-controlled trial in adult psychotic inpatients and outpatients who met the DSM-IV criteria for schizophrenia.
Efficacy data were obtained from 400 patients with schizophrenia who were randomized to receive injections of 25 mg, 50 mg, or 75 mg RISPERDAL CONSTA or placebo every 2 weeks. During a 1-week run-in period, patients were discontinued from other antipsychotics and were titrated to a dose of 4 mg oral RISPERDAL. Patients who received RISPERDAL CONSTA were given doses of oral RISPERDAL (2 mg for patients in the 25-mg group, 4 mg for patients in the 50-mg group, and 6 mg for patients in the 75-mg group) for the 3 weeks after the first injection to provide therapeutic plasma concentrations until the main release phase of risperidone from the injection site had begun. Patients who received placebo injections were given placebo tablets.
Efficacy was evaluated using the Positive and Negative Syndrome Scale (PANSS), a validated, multi-item inventory, composed of five subscales to evaluate positive symptoms, negative symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression.
The primary efficacy variable in this trial was change from baseline to endpoint in the total PANSS score. The mean total PANSS score at baseline for schizophrenic patients in this study was 81.5.
Total PANSS scores showed significant improvement in the change from baseline to endpoint in schizophrenic patients treated with each dose of RISPERDAL CONSTA (25 mg, 50 mg, or 75 mg) compared with patients treated with placebo. While there were no statistically significant differences between the treatment effects for the three dose groups, the effect size for the 75 mg dose group was actually numerically less than that observed for the 50 mg dose group.
Subgroup analyses did not indicate any differences in treatment outcome as a function of age, race, or gender.
Bipolar Disorder - Monotherapy: The effectiveness of RISPERDAL CONSTA for the maintenance treatment of Bipolar I Disorder was established in a multicenter, double-blind, placebo-controlled study of adult patients who met DSM-IV criteria for Bipolar Disorder Type I, who were stable on medications or experiencing an acute manic or mixed episode.
A total of 501 patients were treated during a 26-week open-label period with RISPERDAL CONSTA (starting dose of 25 mg, and titrated, if deemed clinically desirable, to 37.5 mg or 50 mg; in patients not tolerating the 25 mg dose, the dose could be reduced to 12.5 mg). In the open-label phase, 303 (60%) patients were judged to be stable and were randomized to double-blind treatment with either the same dose of RISPERDAL CONSTA or placebo and monitored for relapse. The primary endpoint was time to relapse to any mood episode (depression, mania, hypomania, or mixed).
Time to relapse was delayed in patients receiving RISPERDAL CONSTA monotherapy as compared to placebo. The majority of relapses were due to manic rather than depressive symptoms. Based on their bipolar disorder history, subjects entering this study had had, on average, more manic episodes than depressive episodes.
Bipolar Disorder - Adjunctive Therapy: The effectiveness of RISPERDAL CONSTA as an adjunct to treatment with lithium or valproate for the maintenance treatment of Bipolar Disorder was established in a multi-center, randomized, double-blind, placebo-controlled study of adult patients who met DSM-IV criteria for Bipolar Disorder Type I and who experienced at least 4 episodes of mood disorder requiring psychiatric/clinical intervention in the previous 12 months, including at least 2 episodes in the 6 months prior to the start of the study.
A total of 240 patients were treated during a 16-week open-label period with RISPERDAL CONSTA (starting dose of 25 mg, and titrated, if deemed clinically desirable, to 37.5 mg or 50 mg), as adjunctive therapy in addition to continuing their treatment as usual for their bipolar disorder, which consisted of mood stabilizers (primarily lithium and valproate), antidepressants, and/or anxiolytics. All oral antipsychotics were discontinued after the first three weeks of the initial RISPERDAL CONSTA injection. In the open-label phase, 124 (51.7%) were judged to be stable for at least the last 4 weeks and were randomized to double-blind treatment with either the same dose of RISPERDAL CONSTA or placebo in addition to continuing their treatment as usual and monitored for relapse during a 52-week period. The primary endpoint was time to relapse to any new mood episode (depression, mania, hypomania, or mixed).
Time to relapse was delayed in patients receiving adjunctive therapy with RISPERDAL CONSTA as compared to placebo. The relapse types were about half depressive and half manic or mixed episodes.
Pharmacokinetics: Absorption: Risperdal: Risperidone is well absorbed. The absolute oral bioavailability of risperidone is 70% (CV=25%). The relative oral bioavailability of risperidone from a tablet is 94% (CV=10%) when compared to a solution.
Pharmacokinetic studies showed that RISPERDAL Oral Solution is bioequivalent to RISPERDAL Tablets.
Plasma concentrations of risperidone, its major metabolite, 9-hydroxyrisperidone, and risperidone plus 9-hydroxyrisperidone are dose proportional over the dosing range of 1 to 16 mg daily (0.5 to 8 mg twice daily). Following oral administration of solution or tablet, mean peak plasma concentrations of risperidone occurred at about 1 hour. Peak concentrations of 9‑hydroxyrisperidone occurred at about 3 hours in extensive metabolizers, and 17 hours in poor metabolizers. Steady-state concentrations of risperidone are reached in 1 day in extensive metabolizers and would be expected to reach steady-state in about 5 days in poor metabolizers. Steady-state concentrations of 9-hydroxyrisperidone are reached in 5-6 days (measured in extensive metabolizers).
Food Effect: Food does not affect either the rate or extent of absorption of risperidone. Thus, RISPERDAL can be given with or without meals.
Risperdal Consta: After a single intramuscular (gluteal) injection of RISPERDAL CONSTA, there is a small initial release of the drug (< 1% of the dose), followed by a lag time of 3 weeks. The main release of the drug starts from 3 weeks onward, is maintained from 4 to 6 weeks, and subsides by 7 weeks following the intramuscular (IM) injection. Therefore, oral antipsychotic supplementation should be given during the first 3 weeks of treatment with RISPERDAL CONSTA to maintain therapeutic levels until the main release of risperidone from the injection site has begun [see Dosage & Administration]. Following single doses of RISPERDAL CONSTA, the pharmacokinetics of risperidone, 9-hydroxyrisperidone (the major metabolite), and risperidone plus 9-hydroxyrisperidone were linear in the dosing range of 12.5 mg to 50 mg.
The combination of the release profile and the dosage regimen (IM injections every 2 weeks) of RISPERDAL CONSTA results in sustained therapeutic concentrations. Steady-state plasma concentrations are reached after 4 injections and are maintained for 4 to 6 weeks after the last injection. Following multiple doses of 25 mg and 50 mg RISPERDAL CONSTA, plasma concentrations of risperidone, 9-hydroxyrisperidone, and risperidone plus 9-hydroxyrisperidone were linear.
Deltoid and gluteal intramuscular injections at the same doses are bioequivalent and, therefore, interchangeable.
Distribution: Risperidone is rapidly distributed. The volume of distribution is 1-2 L/kg. In plasma, risperidone is bound to albumin and α1-acid glycoprotein. The plasma protein binding of risperidone is 90%, and that of its major metabolite, 9-hydroxyrisperidone, is 77%. Neither risperidone nor 9‑hydroxyrisperidone displaces each other from plasma binding sites. High therapeutic concentrations of sulfamethazine (100 mcg/mL), warfarin (10 mcg/mL), and carbamazepine (10 mcg/mL) caused only a slight increase in the free fraction of risperidone at 10 ng/mL and 9‑hydroxyrisperidone at 50 ng/mL, changes of unknown clinical significance.
Metabolism and Drug Interactions: Risperidone is extensively metabolized in the liver. The main metabolic pathway is through hydroxylation of risperidone to 9-hydroxyrisperidone by the enzyme, CYP 2D6. A minor metabolic pathway is through N-dealkylation. The main metabolite, 9-hydroxyrisperidone, has similar pharmacological activity as risperidone. Consequently, the clinical effect of the drug results from the combined concentrations of risperidone plus 9-hydroxyrisperidone.
CYP 2D6, also called debrisoquin hydroxylase, is the enzyme responsible for metabolism of many neuroleptics, antidepressants, antiarrhythmics, and other drugs. CYP 2D6 is subject to genetic polymorphism (about 6%-8% of Caucasians, and a very low percentage of Asians, have little or no activity and are "poor metabolizers") and to inhibition by a variety of substrates and some non-substrates, notably quinidine. Extensive CYP 2D6 metabolizers convert risperidone rapidly into 9‑hydroxyrisperidone, whereas poor CYP 2D6 metabolizers convert it much more slowly. Although extensive metabolizers have lower risperidone and higher 9‑hydroxyrisperidone concentrations than poor metabolizers, the pharmacokinetics of risperidone and 9-hydroxyrisperidone combined, after single and multiple doses, are similar in extensive and poor metabolizers.
Risperidone could be subject to two kinds of drug-drug interactions. First, inhibitors of CYP 2D6 interfere with conversion of risperidone to 9-hydroxyrisperidone [see Interactions]. This occurs with quinidine, giving essentially all recipients a risperidone pharmacokinetic profile typical of poor metabolizers. The therapeutic benefits and adverse effects of risperidone in patients receiving quinidine have not been evaluated, but observations in a modest number (n≅70) of poor metabolizers given oral RISPERDAL do not suggest important differences between poor and extensive metabolizers. Second, co-administration of known enzyme inducers (e.g., carbamazepine, phenytoin, rifampin, and phenobarbital) with oral RISPERDAL may cause a decrease in the combined plasma concentrations of risperidone and 9‑hydroxyrisperidone [see Interactions]. It would also be possible for risperidone to interfere with metabolism of other drugs metabolized by CYP 2D6. Relatively weak binding of risperidone to the enzyme suggests this is unlikely [see Interactions].
Risperdal: In vitro studies indicate that risperidone is a relatively weak inhibitor of CYP 2D6. Therefore, RISPERDAL is not expected to substantially inhibit the clearance of drugs that are metabolized by this enzymatic pathway. In drug interaction studies, RISPERDAL did not significantly affect the pharmacokinetics of donepezil and galantamine, which are metabolized by CYP 2D6.
In vitro studies demonstrated that drugs metabolized by other CYP isozymes, including 1A1, 1A2, 2C9, 2C19, and 3A4, are only weak inhibitors of risperidone metabolism.
Risperdal Consta: The interactions of RISPERDAL CONSTA with coadministration of other drugs have not been systematically evaluated in human subjects. Drug interactions are based primarily on experience with oral RISPERDAL.
Excretion: Risperidone and its metabolites are eliminated via the urine and, to a much lesser extent, via the feces. As illustrated by a mass balance study of a single 1 mg oral dose of 14C-risperidone administered as solution to three healthy male volunteers, total recovery of radioactivity at 1 week was 84%, including 70% in the urine and 14% in the feces.
Risperdal: The apparent half-life of risperidone was 3 hours (CV=30%) in extensive metabolizers and 20 hours (CV=40%) in poor metabolizers. The apparent half-life of 9-hydroxyrisperidone was about 21 hours (CV=20%) in extensive metabolizers and 30 hours (CV=25%) in poor metabolizers. The pharmacokinetics of risperidone and 9-hydroxyrisperidone combined, after single and multiple doses, were similar in extensive and poor metabolizers, with an overall mean elimination half-life of about 20 hours.
Risperdal Consta: The apparent half-life of risperidone plus 9-hydroxyrisperidone following RISPERDAL CONSTA administration is 3 to 6 days, and is associated with a monoexponential decline in plasma concentrations. This half-life of 3-6 days is related to the erosion of the microspheres and subsequent absorption of risperidone. The clearance of risperidone and risperidone plus 9‑hydroxyrisperidone was 13.7 L/h and 5.0 L/h in extensive CYP 2D6 metabolizers, and 3.3 L/h and 3.2 L/h in poor CYP 2D6 metabolizers, respectively. No accumulation of risperidone was observed during long-term use (up to 12 months) in patients treated every 2 weeks with 25 mg or 50 mg RISPERDAL CONSTA. The elimination phase is complete approximately 7 to 8 weeks after the last injection.
Specific Populations: Race and Gender Effects: No specific pharmacokinetic study was conducted to investigate race and gender effects, but a population pharmacokinetic analysis did not identify important differences in the disposition of risperidone due to gender (whether or not corrected for body weight) or race.
Risperdal: Renal and Hepatic Impairment: See Precautions.
Elderly: In healthy elderly subjects, renal clearance of both risperidone and 9‑hydroxyrisperidone was decreased, and elimination half-lives were prolonged compared to young healthy subjects. Dosing should be modified accordingly in the elderly patients [see Precautions].
Pediatric: The pharmacokinetics of risperidone and 9-hydroxyrisperidone in children were similar to those in adults after correcting for the difference in body weight.
Risperdal Consta: Renal Impairment: In patients with moderate to severe renal disease treated with oral RISPERDAL, clearance of the sum of risperidone and its active metabolite decreased by 60% compared with young healthy subjects. Although patients with renal impairment were not studied with RISPERDAL CONSTA, it is recommended that patients with renal impairment be carefully titrated on oral RISPERDAL before treatment with RISPERDAL CONSTA is initiated at a dose of 25 mg. A lower initial dose of 12.5 mg may be appropriate when clinical factors warrant dose adjustment, such as in patients with renal impairment [see Dosage & Administration].
Hepatic Impairment: While the pharmacokinetics of oral RISPERDAL in subjects with liver disease were comparable to those in young healthy subjects, the mean free fraction of risperidone in plasma was increased by about 35% because of the diminished concentration of both albumin and α1-acid glycoprotein. Although patients with hepatic impairment were not studied with RISPERDAL CONSTA, it is recommended that patients with hepatic impairment be carefully titrated on oral RISPERDAL before treatment with RISPERDAL CONSTA is initiated at a dose of 25 mg. A lower initial dose of 12.5 mg may be appropriate when clinical factors warrant dose adjustment, such as in patients with hepatic impairment [see Dosage & Administration].
Elderly: In an open-label trial, steady-state concentrations of risperidone plus 9-hydroxyrisperidone in otherwise healthy elderly patients (≥ 65 years old) treated with RISPERDAL CONSTA for up to 12 months fell within the range of values observed in otherwise healthy nonelderly patients. Dosing recommendations are the same for otherwise healthy elderly patients and nonelderly patients [see Dosage & Administration].
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenesis: Oral: Risperidone was administered in the diet at doses of 0.63, 2.5, and 10 mg/kg for 18 months to mice and for 25 months to rats. These doses are equivalent to approximately 0.2, 0.75, and 3 times (mice) and 0.4, 1.5, and 6 times (rats) the MRHD of 16 mg/day, based on mg/m2 body surface area. A maximum tolerated dose was not achieved in male mice. There were statistically significant increases in pituitary gland adenomas, endocrine pancreatic adenomas, and mammary gland adenocarcinomas. The following table summarizes the multiples of the human dose on a mg/m2 (mg/kg) basis at which these tumors occurred. (See Table 1.)
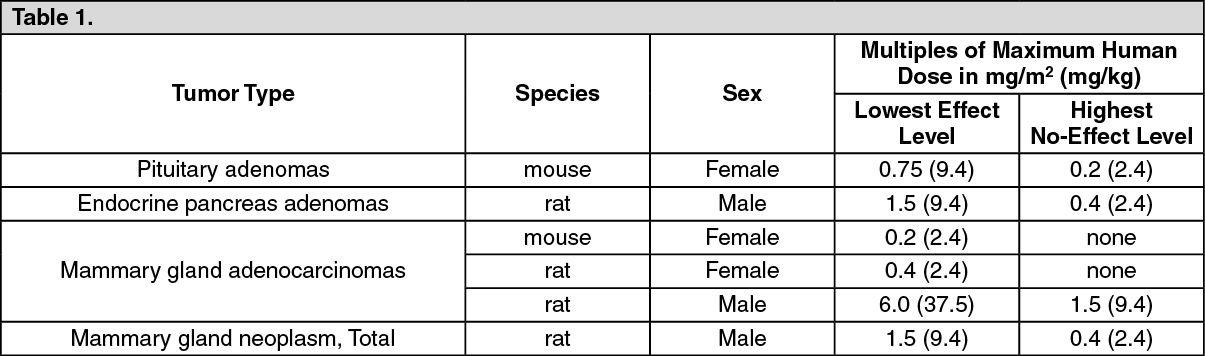 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAntipsychotic drugs have been shown to chronically elevate prolactin levels in rodents. Serum prolactin levels were not measured during the risperidone carcinogenicity studies; however, measurements during subchronic toxicity studies showed that risperidone elevated serum prolactin levels 5-6 fold in mice and rats at the same doses used in the carcinogenicity studies. An increase in mammary, pituitary, and endocrine pancreas neoplasms has been found in rodents after chronic administration of other antipsychotic drugs and is considered to be prolactin‑mediated. The relevance for human risk of the findings of prolactin‑mediated endocrine tumors in rodents is unclear [see Precautions].
Risperdal Consta: Intramuscular: Risperidone was evaluated in a 24-month carcinogenicity study in which SPF Wistar rats were treated every 2 weeks with intramuscular (IM) injections of either 5 mg/kg or 40 mg/kg of risperidone. These doses are 1 and 8 times the MRHD (50 mg) on a mg/m2 basis. A control group received injections of 0.9% NaCl, and a vehicle control group was injected with placebo microspheres. There was a significant increase in pituitary gland adenomas, endocrine pancreas adenomas, and adrenomedullary pheochromocytomas at 8 times the IM MRHD on a mg/m2 basis. The incidence of mammary gland adenocarcinomas was significantly increased in female rats at both doses (1 and 8 times the IM MRHD on a mg/m2 basis). A significant increase in renal tubular tumors (adenoma, adenocarcinomas) was observed in male rats at 8 times the IM MRHD on a mg/m2 basis. Plasma exposures (AUC) in rats were 0.3 and 2 times (at 5 and 40 mg/kg, respectively) the expected plasma exposure (AUC) at the IM MRHD.
Dopamine D2 receptor antagonists have been shown to chronically elevate prolactin levels in rodents. Serum prolactin levels were not measured during the carcinogenicity studies of oral risperidone; however, measurements taken during subchronic toxicity studies showed that oral risperidone elevated serum prolactin levels 5- to 6-fold in mice and rats at the same doses used in the oral carcinogenicity studies. Serum prolactin levels increased in a dose-dependent manner up to 6- and 1.5-fold in male and female rats, respectively, at the end of the 24-month treatment with risperidone every 2 weeks IM. Increases in the incidence of pituitary gland, endocrine pancreas, and mammary gland neoplasms have been found in rodents after chronic administration of other antipsychotic drugs and may be prolactin-mediated.
The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is unknown [see Precautions].
Mutagenesis: No evidence of mutagenic or clastogenic potential for risperidone was found in the in vitro tests of Ames gene mutation, the mouse lymphoma assay, rat hepatocyte DNA-repair assay, the chromosomal aberration test in human lymphocytes, Chinese hamster ovary cells, or in the in vivo oral micronucleus test in mice and the sex-linked recessive lethal test in Drosophila.
Risperdal Consta: In addition, no evidence of mutagenic potential was found in the in vitro Ames reverse mutation test for RISPERDAL CONSTA.
Impairment of Fertility: Oral risperidone (0.16 to 5 mg/kg) impaired mating, but not fertility, in rat reproductive studies at doses 0.1 to 3 times the MRHD of 16 mg/day based on mg/m2 body surface area. The effect appeared to be in females, since impaired mating behavior was not noted in the male fertility study. In a subchronic study in Beagle dogs in which oral risperidone was administered orally at doses of 0.31 to 5 mg/kg, sperm motility and concentration were decreased at doses 0.6 to 10 times the MRHD based on mg/m2 body surface area. Dose-related decreases were also noted in serum testosterone at the same doses. Serum testosterone and sperm parameters partially recovered, but remained decreased after treatment was discontinued. A no‑effect dose could not be determined in either rat or dog.